CASE REPORT | https://doi.org/10.5005/jp-journals-10057-0188 |
Unusual Presentation of Central Neurocytoma in Posterior Part of Third Ventricle Extending into Fourth Ventricle
1-5Department of Radio-Diagnosis, Mahatma Gandhi Medical College & Hospital, Jaipur, Rajasthan, India
Corresponding Author: Vishwajeet A Burungale, Department of Radio-Diagnosis, Mahatma Gandhi Medical College & Hospital, Jaipur, Rajasthan, India, Phone: +91 9561476389, e-mail: vburungale@gmail.com
ABSTRACT
Central neurocytomas (CNs) are usually located in the lateral ventricles. Primary origin in the third ventricle is very rare. We discuss the clinical signs and radiological findings of this tumor. We report a case of a third ventricle CN extending through a cerebral aqueduct in the fourth ventricle in a 40-year-old male patient, who initially presented with progressive headache, vertigo, and blurred vision for 3–4 months. Magnetic resonance imaging of the brain revealed a slightly enhanced tumor in the third ventricle with small necrotic areas, calcification, and obstructive hydrocephalus. Total surgical removal of the tumor was performed. The tumor was diagnosed as a CN on histopathological studies. CNs located in the third ventricle are rare. Total tumor excision is associated with a favorable prognosis.
How to cite this article: Burungale VA, Mishra V, Kathait AS, et al. Unusual Presentation of Central Neurocytoma in Posterior Part of Third Ventricle Extending into Fourth Ventricle. J Mahatma Gandhi Univ Med Sci Tech 2021;6(3):93–96.
Source of support: Nil
Conflict of interest: None
Keywords: Hydrocephalus, Central neurocytoma, Ventricular tumor.
INTRODUCTION
Central Neurocytoma is a low-grade, slow-growing tumor of neuronal origin that was first described by Hassoun et al. in 1982.1 They include 0.25–0.5% of all primary brain tumors.2 CNs are classically situated in the lateral ventricles adjacent to the foramen of Monro.3,4 The primary tumor origin is very rare in the third ventricle. This tumor can spread through cerebrospinal fluid (CSF) and spread along the craniospinal structures and structures outside the central nervous system, such as the peritoneum by means of a ventriculoperitoneal shunt.5,6 CNs are classically seen in patients of age group 20–40 years, and account for not more than 1% of intracranial tumors. There is no testified gender predilection. On CT scan, CNs are generally hyperattenuating related to white matter. Calcification is seen in more than 50% of cases and is typically punctate in nature. The larger tumors usually have cystic regions within them. Generally, there is mild to moderate contrast enhancement, with associated ventricular dilatation. CN appears isointense to grey matter on T1W image and shows mild to moderate heterogeneous enhancement on T1 with contrast images. T2W/FLAIR shows iso- to hyperintense lesion compared to brain with numerous cystic areas (bubbly appearance), many of which are completely attenuated on FLAIR. Prominent flow voids may be seen. GE/SWI images show calcification, typically punctuate in nature. Hemorrhage (especially in larger tumors) is common, uncommonly resulting in ventricular hemorrhage.7
CASE DESCRIPTION
A 40-year-old male patient was admitted to the Mahatma Gandhi Hospital with complaints of progressive headache, vertigo, and blurred vision for 3–4 months. The headaches were more severe in the morning. The patient presented with normal neurological status. The physical examination revealed no abnormalities. The neurological examination demonstrated mild papilledema, that was suggestive of increased intracranial pressure.
Magnetic resonance imaging (MRI) of the patient was done. T1, T2, FLAIR, SW images of brain were obtained in multiple planes. Postcontrast T1 images were also obtained in multiple planes. MRI revealed a lobulated mass lesion with restricted diffusion within the posterior part of third ventricle extending into the cerebral aqueduct and proximal part of fourth ventricle measuring about 45 × 28 × 46 mm (AP × TD × CC) (Figs 1 and 4). The lesion showed lobulated margins with small tiny cystic/necrotic areas within it. Very small calcifications were also seen within the lesion on SW images (Fig. 5). Lesion was seen bulging into the pineal gland region and extending into the supracerebellar cistern mildly effacing the adjacent cerebellar parenchyma. There was mass effect over midbrain and pons as well. Splaying of tectal plate was observed. Lesion was seen to be causing marked obstructive dilatation of supratentorial ventricular system with mild periventricular ooze. On postcontrast study, the lesion showed mild enhancement (Fig. 2). There was also increased amount of fluid along bilateral optic nerve sheaths-suggestive of raised intracranial tension. Based on imaging findings, differential diagnoses of lesion were CN, ependymoma, glioma, and germinoma. Germinomas are usually in the age group of 10–20 years and areas of necrosis are not seen. Ependymomas are usually well defined and homogenous with dense enhancement on post-gadolinium (Gd)-scan. So, our diagnosis was either a CN or an ependymoma.

Figs 1A and B: T2W axial and sagittal images showing mildly hyperintense lobulated lesion within the posterior aspect of the third ventricle extending into the cerebral aqueduct and fourth ventricle and mildly bulging into the pineal gland region posteriorly with a few small cystic areas within it. Ventricular system appears dilated

Figs 2A and B: On postcontrast FAT SAT T1W coronal and sagittal images lesion is showing moderate homogeneous enhancement
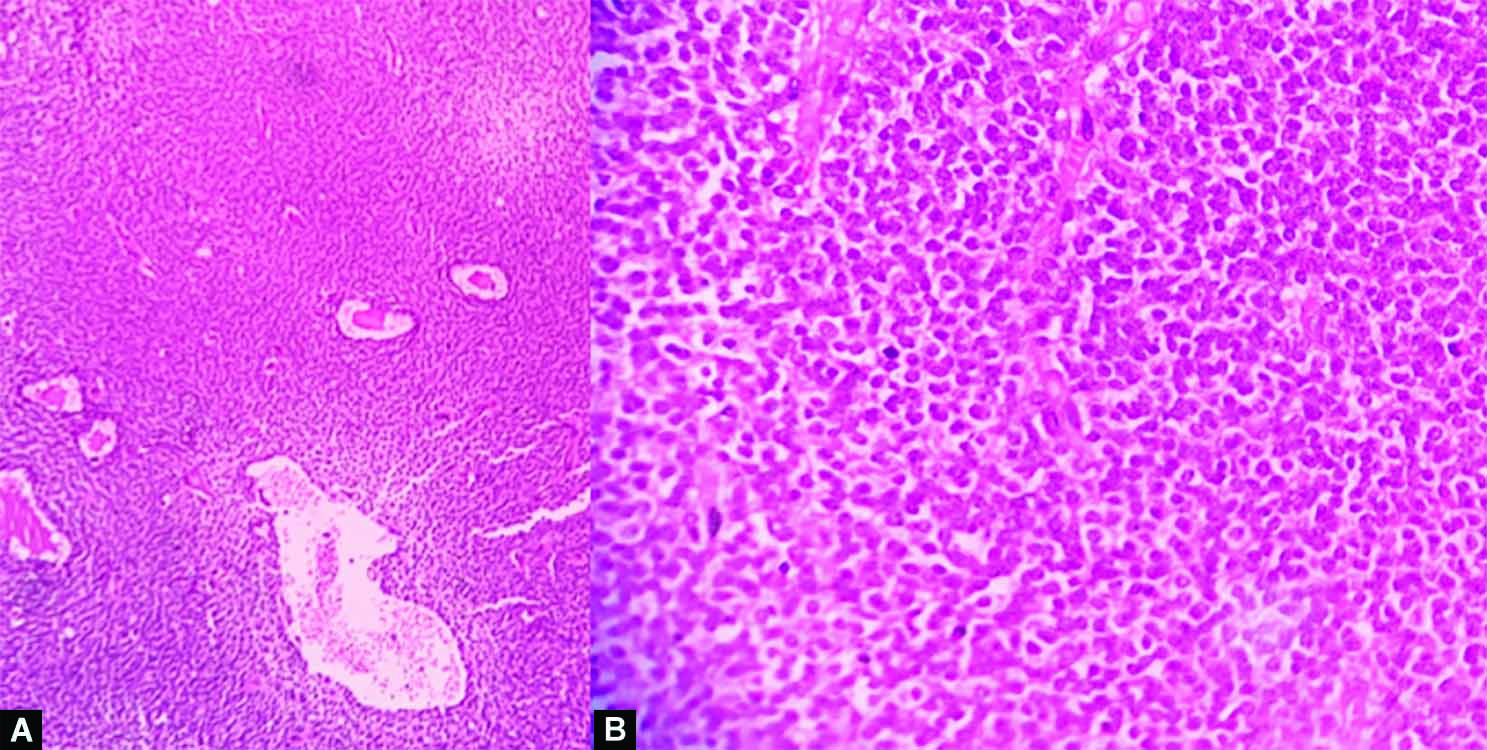
Figs 3A and B: Histopathology correlation showed cells having uniform round to oval nuclei, finely speckled chromatin, and variably present nucleoli arranged in sheets. Occasional rosette-like areas were seen

Figs 4A and B: Lesion is showing restricted diffusion appearing hyperintense on DWI image and hypointense on corresponding ADC map
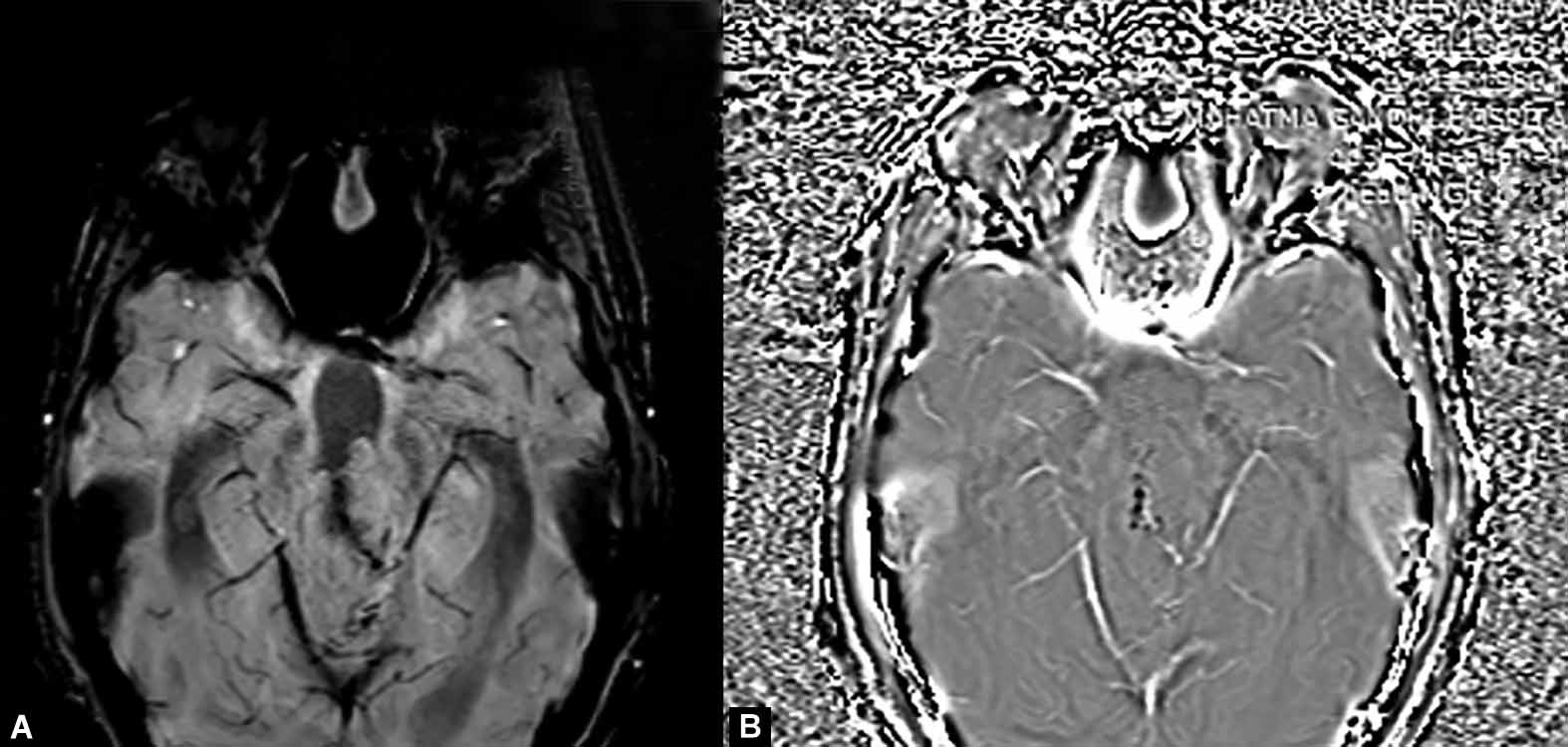
Figs 5A and B: Lesion is showing evidence of minimal calcification appearing hypointense on SW and phase images
Based on these findings, the patient underwent a midline sub-occipital craniotomy. A soft, grayish, vascular mass attached to the posterior part of third ventricle was recognized and completely removed via a transvermis approach. The consistency of the tumor was soft and suckable. The histological examination of the surgical specimen depicted that the tumor comprised of cells having uniform round to oval nuclei, finely speckled chromatin, and unevenly present nucleoli arranged in sheets. Occasional rosette-like areas were also seen. Blood vessels were thin walled. Mitotic activity was not significant. The final pathological diagnosis confirmed a central neurocytoma (WHO Grade II).
In course of follow-up, headache and papilledema of the patient were settled after the operation. The patient had no significant complaint. CN in third ventricle is very rare and only consists of around 5% of total neurocytomas. CN which is arising from the third ventricle and going into the fourth ventricle has not been reported yet in our knowledge. The enhancement pattern of our case was also different than normal neurocytomas (Fig. 3).
DISCUSSION
Intraventricular CNs are rare tumors of neuroepithelial origin, show incidences opening from 0.1–0.5% and are altogether a large series of central system nervosum neoplasms.8,9 Young adults are mainly affected by the CN, with a mean age of roughly 30 years and both males and females are equally affected.8,10 The typical site of tumor origin is the lateral ventricle, with most common location being adjacent to the foramen of Monro. The neurocytoma cells may arise from remnants of the subependymal plate or from nuclei of gray matter within the septum pellucidum and ventricle8,11,12 and this hypothesis may clarify the intraventricular site of CNs. Maximum patients show symptoms of raised intracranial pressure induced by obstructive hydrocephalus or mass effects induced by the tumor, like headaches, vomiting, diplopia, blurred vision, and memory impairment. Papilledema is seen in about half of patients at the time of diagnosis. The typical time duration of clinical symptoms is nearly 6 months.8,10 Our patient presented with a comparatively shorter clinical history owing to the critical location of the tumor within the posterior fossa. CNs normally present with an inhomogeneous signal in T1-weighted resonance imaging scans; whereas the signal in T2-weighted resonance imaging scans is variable. Hydrocephalus may be a common finding. The MRI findings for our patient resembled those described above. As total tumor excision is related to a favorable prognosis, MRI is an extremely important tool in diagnosis as well as getting to know the surgical extent of the tumor.
In light-microscopic analysis, CNs display small uniform cells with perinuclear halos and perivascular pseudo-rosettes; hence, it is tough to distinguish CNs from oligodendrogliomas and ependymomas. Immunohistochemical staining for markers like synaptophysin, neuron-specific enolase (NSE) and glial fibrillary acidic protein (GFAP) can offer a valuable distinction tool. Positive synaptophysin and neuron-specific enolase staining depict a neuroepithelial cell origin, and negative glial fibrillary acidic protein staining claims against glial differentiation. All these findings established our diagnosis of CN in this patient. Transmission microscopy can be beneficial when tumor marker expression is unclear. CNs reveal features of neuronal differentiation like prominent mitochondria, Golgi body, microtubules, thin cell processes, dense-core neurosecretory vesicles and well-formed synapses.4,6,11,12
The clinical progression of CNs is classically indolent, and they are classified histologically as WHO grade II. Complete surgical excision can enable long-term local control of the tumor.13,14 Atypical CNs, as first noted by Soylemezoglu et al.,15 are known to show Ki-67/MIB-1 indices and/or vascular proliferation, mitosis, and necrosis15 and demonstrate significantly shorter recurrence-free intervals. For control of tumor, postoperative radiotherapy with a minimal dose of 5000 cGy is used.8,10,13
CONCLUSION
CNs are rare intraventricular tumors of neuronal origin, which are characteristically slow-growing in nature. Typical MR findings include a predominantly solid portion with intratumoral cystic changes and broad attachment to the septum pellucidum and/or lateral wall of the ventricle showing restricted diffusion and enhancement. Although they are very rare, ventricular CNs must be kept in mind. The critical location within the ventricle may explain a shorter clinical history. Total tumor excision can produce a satisfactory prognosis. Postoperative radiotherapy could even be considered for cases of subtotal excision, anaplastic histological variants, or recurring tumors. MRI is beneficial in defining tumor extension, which is significant in preoperative planning.
REFERENCES
1. Hassoun J, Gambarelli D, Grisoli F, et al. Central neurocytoma: an electron–microscopic study of two cases. Acta Neuropathol 1982;56(02):151–156. DOI: 10.1007/BF00690587
2. Hassoun J, Söylemezoglu F, Gambarelli D, et al. Central neurocytoma: a synopsis of clinical and histological features. Brain Pathol 1993;3(03):297–306. DOI: 10.1111/j.1750-3639.1993.tb00756.x
3. Ishiuchi S, Tamura M. Central neurocytoma: An immunohistochemical, ultrastructural and cell culture study. Acta Neuropathol 1997;94(05):225–435. DOI: 10.1007/s004010050729
4. Majos C, Coll S, Pons LC. Central neurocytoma arising within the ventricle. Neuroradiology 1997;39(4):270–272. DOI: 10.1007/s002340050406
5. Takao H, Nakagawa K, Ohtomo K. Central neurocytoma with craniospinal dissemination. J Neurooncol 2003;61(3):255–259. DOI: 10.1023/a:1022553110232
6. Buccoliero AM, Caldarella A, Ammannati F, et al. Extraventricular neurocytoma: morphological and immunohistochemistry considerations on medical diagnosis. Pathol Res Pract 2002;198(9):627–633. DOI: 10.1078/0344-0338-00312
7. Smith A, Smirniotopoulos J, Horkanyne-Szakaly I, et al. From the radiologic pathology archives: intraventricular neoplasms: radiologic-pathologic correlation. Radiographics 2013;33(01):21–43. DOI: 10.1148/rg.331125192.
8. Drevelengas A, Polyzoides K, Kalaitzoglou I. Intraventricular neurocytoma: case report and review. Eur J Radiol 1994;19(1):14–18. DOI: 10.1016/0720-048X(94)00555-q
9. Park S-H, Ostrzega N, Akers M-A, et al. Intraventricular neurocytoma with prominent myelin figures. Ultrastruct Pathol 1999;23(5):311–317. DOI: 10.1080/019131299281455
10. Agranovich AL, Ang L-C, Fryer CJH. Central neurocytoma: report of 2 cases and literature review. J Neurooncol 1993;16(1):47–53. DOI: 10.1007/BF01324834
11. Robbins P, Segal A, Narula S, et al. Central neurocytoma: a clinicopathological, immunohistochemical and ultrastructural study of seven cases. Pathol Res Pract 1995;191(2):100–111. DOI: 10.1016/S0344-0338(11)80559-5
12. Warmuth-Metz M, Klein R, Sorensen N, et al. Central neurocytoma of the fourth ventricle. J Neurosurg 1999;91(03):506–509. DOI: 10.3171/jns.1999.91.3.0506
13. Mackenzie IRA. Central neurocytoma: histologic atypia, proliferation, and clinical outcome. Cancer 1999;85(07):1606–1610. DOI: 10.1002/(sici)1097-0142(19990401)85:7<1606::aid-cncr24>3.0.co;2-b
14. Ng TH, Wong AY, Compton JS, et al. Pigmented central neurocytoma: case report and literature review. Am J Surg Pathol 1999;23(9):1136–1140. DOI: 10.1097/00000478-199909000-00019
15. Soylemezoglu F, Scheithauer BW, Esteve J, et al. Atypical central neurocytoma. J Neuropathol Exp Neurol 1997;56(5):551–556. DOI: 10.1097/00005072-199705000-0001110.1097/00005072-199705000-00011
________________________
© The Author(s). 2021 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.